9 热图
9.1 什么是热图?
在组学研究的相关文章中,我们常常可以看到热图(Heatmap)的展示。这些红绿相间且色彩变化丰富的热图总是能吸引读者的眼球,从而为文章增添不少亮色。当然,作为严谨的科学研究论文,图表的展示当然不可能仅仅是为了好看。热图作为一种对实验数据及其分析结果的直观的表达方式,在很多文章中都有着不可或缺的地位。
它是一种将规则化矩阵数据转换成颜色色调的常用的可视化方法,其中每个单元格对应数据的某些属性,属性的值通过颜色映射转换为不同色调并按规则填充单元格。
本文我们就来讨论一下热图是如何绘制的以及如何对其进行解读。
9.2 绘图前的数据准备
demo数据可以在https://www.bioladder.cn/shiny/zyp/demoData/heatmap.rar下载。
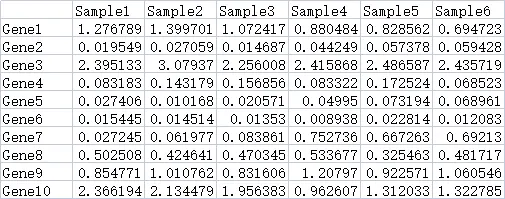
9.2.1 热图数据
数据来源一般是搜库结果定量表。包含2个维度的数据,一般情况下,每一行是一个基因,每一列是一个样本。
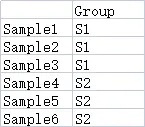
9.2.2 样本分组数据(可选)
行名的名称和个数要和之前的heatmap数据保持一致,列名为分组名称,可以包含不止一个分组。
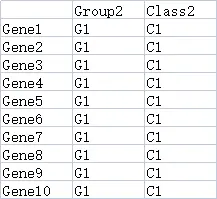
9.2.3 基因分组数据(可选)
行名的名称和个数要和之前的heatmap数据保持一致,列名为分组名称,可以包含不止一个分组。
9.3 R语言怎么画热图
library(pheatmap) # 加载pheatmap这个R包
# 1,读取热图数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/demoData/heatmap/data.heatmap.txt", #文件名称 注意文件路径,格式
header = T, # 是否有标题
sep = "\t", # 分隔符是Tab键
row.names = 1, # 指定第一列是行名
fill=T) # 是否自动填充,一般选择是
# (可选)读取分组数据文件
# dfSample = read.delim("https://www.bioladder.cn/shiny/zyp/demoData/heatmap/sample.class.txt",header = T,row.names = 1,fill = T,sep = "\t")
# dfGene = read.delim("https://www.bioladder.cn/shiny/zyp/demoData/heatmap/gene.class.txt",header = T,row.names = 1,fill = T,sep = "\t")
# 2,绘图
pheatmap(df,
# annotation_row=dfGene, # (可选)指定行分组文件
# annotation_col=dfSample, # (可选)指定列分组文件
show_colnames = TRUE, # 是否显示列名
show_rownames=TRUE, # 是否显示行名
fontsize=2, # 字体大小
color = colorRampPalette(c('#0000ff','#ffffff','#ff0000'))(50), # 指定热图的颜色
annotation_legend=TRUE, # 是否显示图例
border_color=NA, # 边框颜色 NA表示没有
scale="row", # 指定归一化的方式。"row"按行归一化,"column"按列归一化,"none"不处理
cluster_rows = TRUE, # 是否对行聚类
cluster_cols = TRUE # 是否对列聚类
)
# 更多参数可以输入 ?pheatmap 查看9.4 BioLadder生信云平台在线绘制热图
不想写代码?可以用BioLadder生信云平台在线绘制热图。
免费使用,注册登录后畅享40+模块。
网址:https://www.bioladder.cn/web/#/chart/6
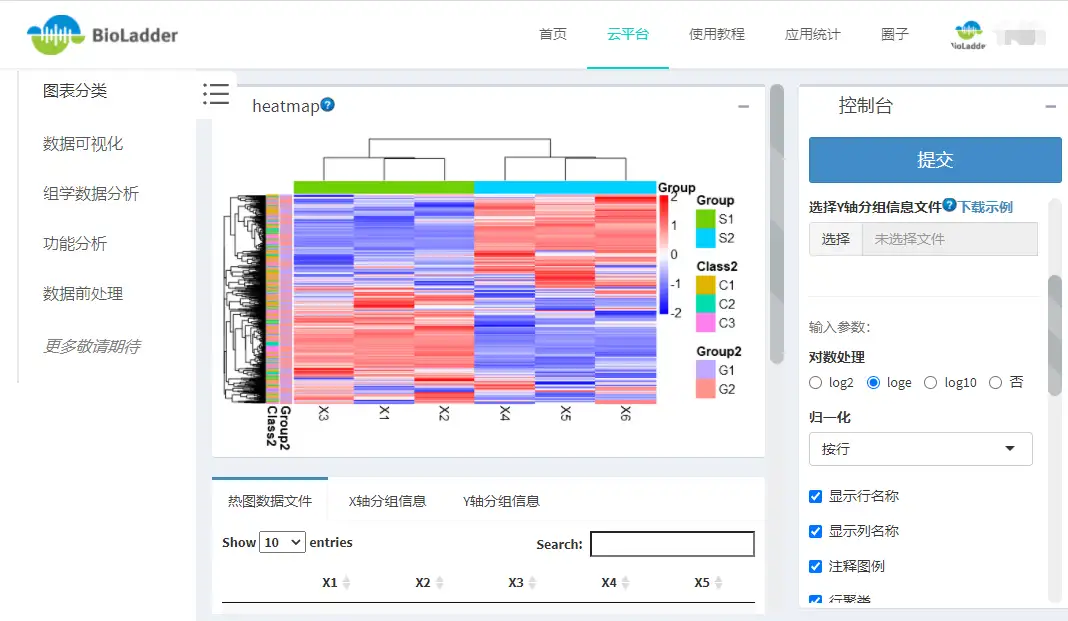
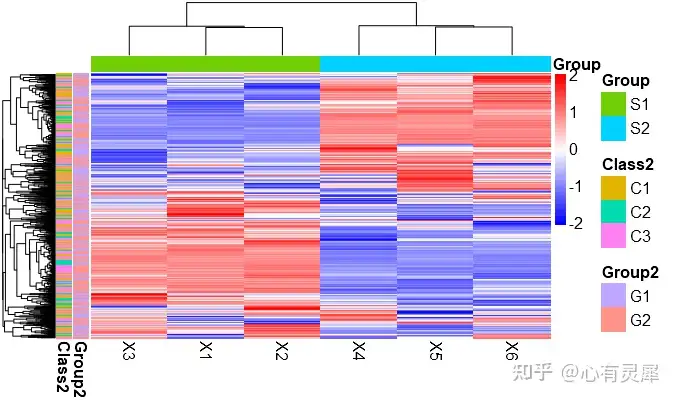
9.5 热图怎么看?
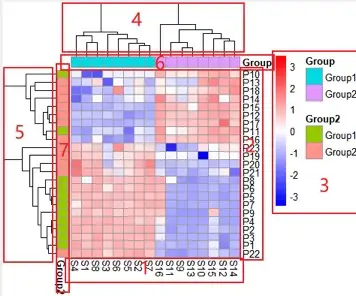
如图,就是一副组学研究中热图的常用绘制模式,每个小方格表示每个基因在不同样本中的定量值,其颜色表示该基因表达量大小,表达量越大颜色越深(红色为高表达,蓝色为低表达)。每行表示每个基因在不同样本中的表达量情况,每列表示每个样品中所有基因的表达量情况。上方树形图表示对来自不同实验分组的不同样品的聚类分析结果,左侧树状图表示对来自不同样本的不同基因的聚类分析结果。
1,行名称,一般为分组名称
2,列名称,一般为基因名称
3,图例信息,左侧是热图小方块的颜色图例说明,右侧为分组信息的图例说明。
4,列聚类,如果不聚类,排序将保持文件数据的默认方式。从样本角度讲,聚类可以观察到你采集的不同组别样本是否被分类到一起了。因为,理论上如果样本来自于同一个组,其特征应该是相似的,而如果在实际操作中,某一个应该属于该组的样本被聚类到别的组了,那就说明这个样本本身的变异度很高,或者说在之前的样本采集或者测序过程中出了什么问题。
5,行聚类,如果不聚类,排序将保持文件数据的默认方式。从基因表达角度讲,聚类可以观察到哪些基因群体具有比较一致的表达变化,因为基因的上下游关系一般是连锁反应的,也就是说一个基因的表达增加可能能够带动一系列的基因的表达增加。
6,列分组信息。
7,行分组信息。